The characterization of aggregation-prone protein variants is essential to understanding the etiology of protein-aggregation diseases and also to the identification of solubility determinants inherent to protein sequences. However, structure determination of these insoluble, usually non-crystalline aggregates represent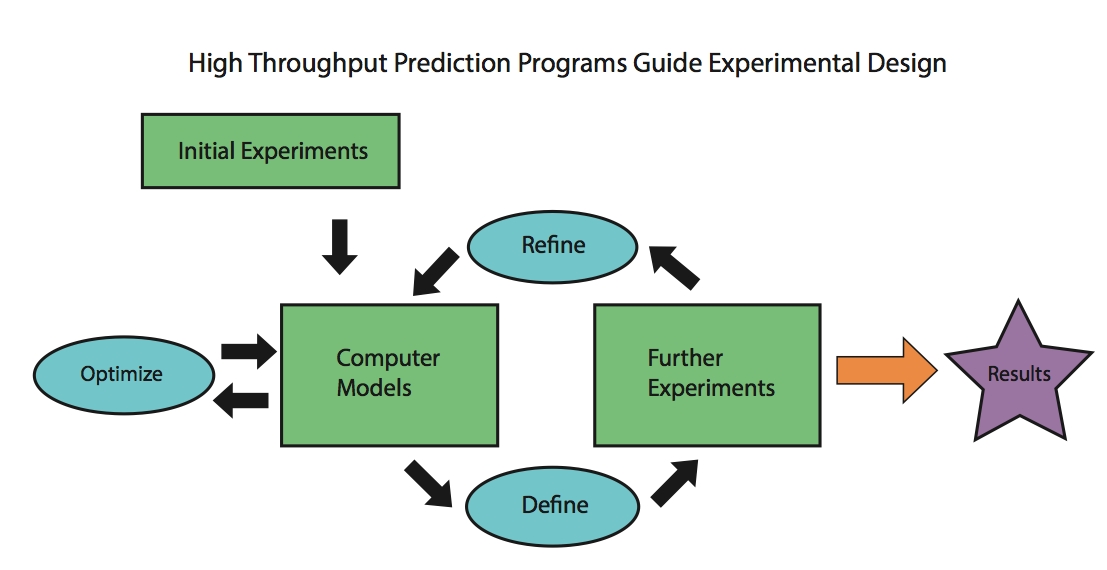 s a major research effort, and even relatively inexpensive techniques can become problematic in a high-throughput context. Prediction of differences in relative solvent accessibility (RSA) between wild-type and variant proteins provides a starting point for investigating potential contacts between surface residues on copies of the same protein that lead to aggregation. Our focus is on the use of Bayesian model integration methods to predict whether there is a change in the surface exposure of any residues upon introducing a single point mutation, which we use as an indirect predictor of increased aggregation propensity, as the sequestration of hydrophobic residues from the surface is necessary for preventing aggregation. Because predictions regarding changes in exposed hydrophobic surface area can be readily verified by relatively inexpensive assays, candidate variants generated by the initial analysis can be experimentally screened prior to further analysis. Bayesian statistical methods in have heretofore been under-explored in the field of bioinformatics, and they have the advantage of allowing for the coupling of prior knowledge of a system with the data sets. Accurate predictions would give valuable information about protein structure and will be used to guide future experimental studies.
s a major research effort, and even relatively inexpensive techniques can become problematic in a high-throughput context. Prediction of differences in relative solvent accessibility (RSA) between wild-type and variant proteins provides a starting point for investigating potential contacts between surface residues on copies of the same protein that lead to aggregation. Our focus is on the use of Bayesian model integration methods to predict whether there is a change in the surface exposure of any residues upon introducing a single point mutation, which we use as an indirect predictor of increased aggregation propensity, as the sequestration of hydrophobic residues from the surface is necessary for preventing aggregation. Because predictions regarding changes in exposed hydrophobic surface area can be readily verified by relatively inexpensive assays, candidate variants generated by the initial analysis can be experimentally screened prior to further analysis. Bayesian statistical methods in have heretofore been under-explored in the field of bioinformatics, and they have the advantage of allowing for the coupling of prior knowledge of a system with the data sets. Accurate predictions would give valuable information about protein structure and will be used to guide future experimental studies.