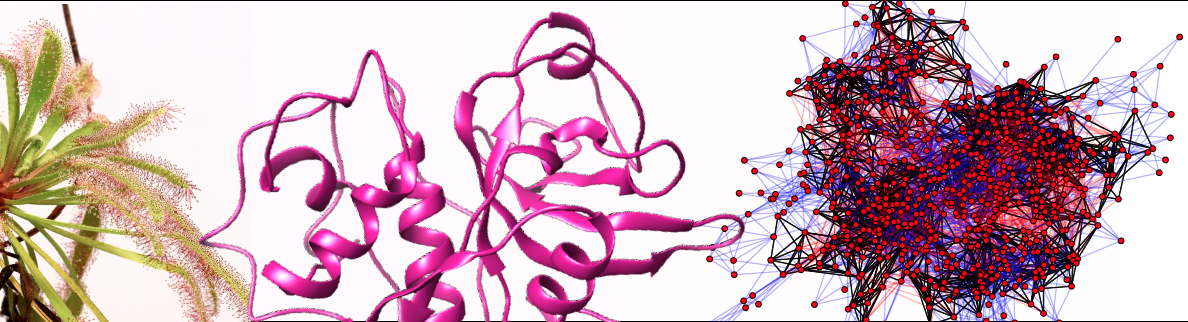
 With our collaborator Carter Butts (Sociology, Statistics and EECS), we have recently sequenced the genome of the carnivorous plant Drosera capensis. This represents the first carnivorous plant genome from order Caryophyllales, and only the third genome of any carnivorous plant. The order Caryophyllales include some of the most specialized examples of plant carnivory, including the tropical pitcher plants (Nepenthes sp.), the Venus flytrap (Dionaea muscipula), and the sundews (Drosera sp.). However, no genome of a carnivorous plant from order Carophylalles has previously been sequenced, limiting the ability of researchers to discover novel enzymes from these sources. Using the genomic “source code,” we have found discovered dozens of novel protease sequences, including pepsin-like aspartic proteases whose existence was first hypothesized by Darwin in 1875.
With our collaborator Carter Butts (Sociology, Statistics and EECS), we have recently sequenced the genome of the carnivorous plant Drosera capensis. This represents the first carnivorous plant genome from order Caryophyllales, and only the third genome of any carnivorous plant. The order Caryophyllales include some of the most specialized examples of plant carnivory, including the tropical pitcher plants (Nepenthes sp.), the Venus flytrap (Dionaea muscipula), and the sundews (Drosera sp.). However, no genome of a carnivorous plant from order Carophylalles has previously been sequenced, limiting the ability of researchers to discover novel enzymes from these sources. Using the genomic “source code,” we have found discovered dozens of novel protease sequences, including pepsin-like aspartic proteases whose existence was first hypothesized by Darwin in 1875.
The proteases of carnivorous plants present attractive targets for exploitation in chemical biology and biotechnology. Carnivorous plants, such as the Cape Sundew (D. capensis), have been rigorously selected by evolution for the ability to digest large prey over long time periods, without assistance from physical disruption of prey tissue, and in competition with ubiquitous fungi and bacteria. These evolutionary constraints have selected for highly stable enzymes that function over a range of pH values and temperatures, with a different profile of substate specificities and cleavage patterns than those found in animal digestive enzymes. Characterization of these proteins will allow their use in a variety of laboratory and applications contexts, including analytical use in proteomics as well as preventing fouling on the surface of medical devices that cannot be treated under harsh conditions. New proteases may also prove useful for cleaving amyloid fibrils, such as those responsible for the transmission of prion diseases or the formation of biofilms by pathogenic bacteria. The characterization of aspartic proteases from the tropical pitcher plants (Nepenthes sp.) [1,2,3], has already led to useful advances in mass spectrometry-based proteomics applications, where the ability to digest proteins using a variety of cut sites is essential for identifying proteins and peptides from complex mixtures. Proteases from plant and animal sources are also important components of pharmaceutical preparations for gluten intolerance, arthritis, and pancreatic disease. Characterizing proteases from carnivorous plants can potentially greatly diversify the toolbox of proteases with different functional properties that are available for these and other applications.
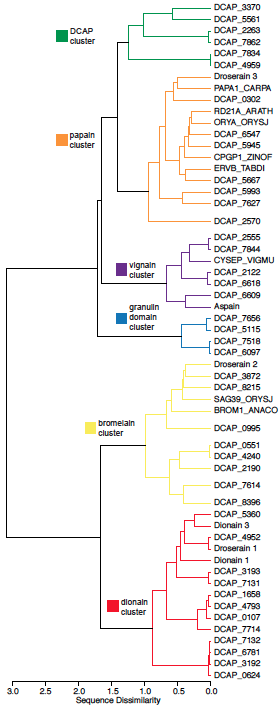
Using Illumina technology, we have have sequenced and assembled a draft genome for D. capensis and identified gene sequences for 44 cysteine proteases and 7 aspartic proteases, all previously unknown. Although active site residues are conserved, the sequence identity of these proteases to known proteins is moderate to low; therefore, comparative modeling with all-atom refinement and subsequent atomistic MD-simulation is used to predict their 3D structures. This in silico methodology provides a general framework for investigating a large pool of sequences that are potentially useful for biotechnology applications, enabling informed choices about which proteins to investigate in the laboratory. We believe this approach illustrates a generally applicable way to leverage the wealth of information provided by whole genome shotgun sequencing for proteomics: computational techniques, despite their limitations, are now powerful enough to allow potentially useful proteins to be identified directly from the genome (especially when backed up by judicious use of homology and comparative analysis with data from related organisms). Given the rapid proliferation of genomic data, being able to identify and pre-check good targets before beginning experimental characterization is a major concern. The immediate aim of this project is to follow up on our in silico studies in order to characterize these novel proteases and harness their unique functionalities for proteomics, chemical biology and biomedical applications.
This approach represents a new road map for rich annotation of genomic data. This is a generally applicable method for exploiting the wealth of information provided by whole genome shotgun sequencing for proteomics: computational techniques, despite their limitations, are now powerful enough to allow potentially useful proteins to be identified directly from genomic “source code” and filtered for strong indicators of biochemical function. Given the rapid proliferation of genomic data, the ability to identify and pre-check good targets for use in chemical biology applications is a major concern. These papers demonstrate how state of the art computational tools can greatly expedite the process of going from sequence data to promising expression candidates.